

原子力システム 研究開発事業 成果報告会資料集
電解還元法を適用した酸化物燃料の乾式再処理に関する技術開発
(研究代表者)坂村義治 原子力技術研究所
(再委託先)独立行政法人日本原子力研究開発機構、国立大学法人京都大学
1.研究開発の背景とねらい
金属燃料高速炉と乾式再処理を組み合わせた金属燃料サイクルは、将来の高速増殖炉システムとして高い性能を有している。一方、軽水炉の燃料は酸化物であるため、将来、金属燃料サイクルに移行していくためには、軽水炉サイクルの酸化物燃料を原料として受け取り、金属形態に還元して金属燃料サイクルに供給するプロセスが必要である。簡素なプロセスが特徴である電解還元法は、その最有力候補に挙げられている。
図1に、電解還元法を適用した酸化物燃料の再処理プロセスの概要を示した。「①前処理」では、使用済燃料を脱被覆して電解還元用の陰極容器に装荷するが、その過程で、適切な形態に成型すること、高温処理によりセシウム(Cs)など一部の核分裂生成物(FP)を揮発分離することも必要に応じて行う。「②電解還元処理」の原理は、次式で表される。
陰極:金属への還元 MO2 + 4 e- → M + 2 O2- … (1)
陽極:酸素ガス発生 2 O2- → O2 + 4 e- … (2)
LiCl溶融塩中での電解により、陰極に装荷された酸化物から酸素がイオンとなって溶融塩中に溶出し、酸化物は金属に還元される。一方、陽極では酸素ガスが放出される。マイナーアクチニド元素(MA)は、ウラン(U)やプルトニウム(Pu)と共に金属に還元されて回収され、FPの内、Csやストロンチウム(Sr)は溶融塩中に溶解して分離される。「③電解精製処理」は、電解還元処理で回収された還元物から残留するFPを取り除くと共に、Pu富化度を調整するために行う。
陽極:金属の溶解 M → M3+ + 3 e- … (3)
固体陰極:ウランの選択的析出 U3+ + 3 e- → U … (4)
液体カドミウム陰極:U,Pu,MAの一括析出 M3+ + 3 e- → M(Cd) … (5)
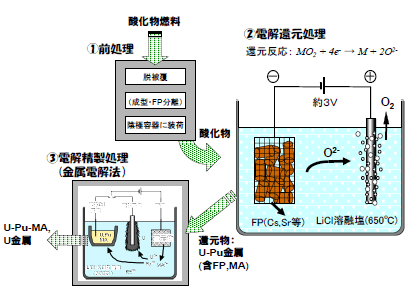
図1 電解還元法を適用した酸化物燃料の乾式再処理プロセス
ここでは、LiCl-KCl溶融塩中で還元物を陽極として電解し、固体陰極でU金属を、液体カドミウム陰極でU-Pu-MA合金を回収する。その過程で、不純物のFPは陽極に残留あるいは溶融塩中に溶解して分離される。
電解還元処理の研究において、これまでにU,Pu,MAが酸化物から金属に還元されることが実証されている1-3)。この電解還元を再処理技術として実用化するためには、酸化物の還元速度を向上させると共に、前処理/電解還元/電解精製の一連プロセスを工学的観点から実証すること、MAやFPの挙動を詳細に把握してプロセスの最適化を図ること、実用的な陽極を開発することが現段階では重要である。本研究開発では、それらの課題に取り組み、次段階である使用済燃料試験や実規模試験に繋げて行く。
2.研究開発成果
2.1.電解還元処理における酸化物還元速度の向上
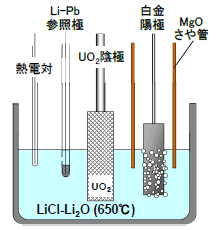
図2 電解還元槽の概略
電解還元技術を実用化するためには、処理速度が十分に大きいことが不可欠である。本研究開発では、実規模として想定している1バッチ当り5〜10 kgの酸化物を、10時間以内で金属に還元できる技術を確立することが1つの目標である。そこで、還元速度向上の観点から、酸化物を装荷する陰極容器の形状と、酸化物原料の形態について検討した。
還元試験で用いた電解槽の概略を図2に示す。温度は650℃で、LiClに約1 wt%のLi2Oを加えた溶融塩を直径10 cmのステンレス製るつぼに入れ、中央にUO2を装荷したステンレス製の陰極バスケットを、その周囲に3つの白金陽極を配置した。陽極の白金板は周囲をマグネシアさや管で囲い、電解中に発生する酸素ガスを上部から排気した。
図3(a)に、90 gのUO2粉末を装荷した陰極バスケットの写真を示した。直径28 mmの皿型バスケットを3段に積み重ねた形状である。電解は15 Aで開始して、還元の進行に伴って設定電流を徐々に下げ、8.4時間後に終了した(終了点は、陰極開放電位から判断)。電解後、陰極バスケットを切断して観察したところ(図3(b))、UO2は完全に金属ウランへ還元されていた。バスケット形状を変化させた試験の結果から、装荷されたUO2粉末の層が厚い場合には、完全に還元するまでに長時間を要することが判明し、図3(a)のバスケット形状が適切であることが示された。バスケット径を大きくし、かつ段数を増やせば、実規模に拡張することは容易であると考えられる。
酸化物原料形態に関する試みとして、あらかじめ多孔質性のUO2ペレットを焼結し、円筒形の陰極バスケットに104 g装荷した(図4(a))。ペレットの寸法は直径7 mm×高さ10 mmで、気孔率は30%である。電解は、同様に15 Aで開始して、9.3時間後に終了した。図4(b)に電解後のバスケット内とペレット断面を示したが、全てのUO2ペレットは金属に還元されていた。多孔質性のUO2ペレットは、ペレット内部に溶融塩が浸透しやすいこと、目の粗い金網で陰極容器を作製できること、バスケット内に溶融塩の流路が確保されることなどから、溶融塩を介した酸素の排出が促進され、酸化物還元速度の向上に効果的であることが示された。また、還元速度はペレット個々の還元速度に律速され、総装荷量には依存しないことも確認された。
以上、実規模で酸化物を10時間以内に還元するという目標を達成できる見込みが示された。

図3 UO2粉末の電解還元試験

図4 多孔質性UO2ペレットの電解還元試験
2.2 模擬使用済燃料を用いた再処理プロセス連続試験
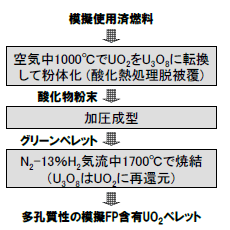
図5 前処理の試験手順
図1に示す前処理/電解還元/電解精製の再処理プロセスを、模擬使用済燃料を約100 g/バッチで用いて、連続的に試験した。前処理の手順を図5に示す。酸化熱処理脱被覆法(酸化物燃料を粉体化して被覆管から分離)を想定し、出発物質の模擬使用済燃料(代表的FP元素のCe,Sr,Nd,Sm, Zr,Mo,Pdを各1 wt%含むUO2ペレット)を、まず空気中・1000℃で酸化し、ウランをU3O8に転換して粉末を得た。次に粉末を加圧成型し、N2-13%H2気流中・1700℃で焼結・還元することにより、気孔率32%の多孔質性酸化物ペレットを作製した(図6)。この簡易なペレット化により、前述した還元反応の促進に加えて、以降のプロセスで微粉末の取扱いを回避できること、焼結の過程で電解還元に悪影響を及ぼすFP(Cs, Te等)を揮発除去できる利点も大きい。
100.8 gの酸化物ペレットを図4(a)と同様の陰極バスケットに入れ、図2の電解還元槽を用いて電解を7.8 時間行った。設定電流は、開始時の15 Aから還元の進行に伴って1 Aまで徐々に下げた。電解後の生成物はペレット形状を保持しており、断面観察の結果、ごく一部を除いてUO2は全て金属に還元されていた(図7)。還元率は99.2%、電流効率は74%、生成物への塩付着率は18 wt%と評価された。FP元素では、
Srは全量が塩中に溶出した。MoとPdは金属、Zrは金属あるいは酸化物として還元生成物中に存在することが分かった。希土類(Ce,Nd,Sm)の酸化物は、一部がPdと合金化して還元され、一部が塩中に溶出した。
電解精製槽の概略を図8に示した。電解還元の生成物を陽極バスケットに装荷し、ステンレス棒を陰極として、500℃の溶融LiCl-KCl-3.5wt%UCl3中で、電流制御による電解を6.0 時間行った。設定電流は、開始時の6 Aから陽極での金属ウラン溶解に伴って下げて行き、0.3 A まで低下した時点で終了した。電解後の陰極には、金属ウランが樹枝状に析出していた(図9(a))。陽極バスケット内には、電解前のペレット形状を保持した残留物が存在していたが(図9(b))、分析の結果、それらの大部分は塩であり、金属ウランの残留率は装荷量の7.4%、電流効率は77%と評価された。なお、実プロセスでは還元生成物を注ぎ足しながら、電解を効率的に進めていく。FP 元素については、希土類元素は塩中に溶出し、Zr,Mo,Pdは大部分が陽極残留物中に存在していた。
以上、実用的な処理速度として目標に掲げていた10 時間以内で酸化物を還元し、さらに精製された金属ウランを高い物質収支で回収することができた。FP 元素の挙動は、概ね予想通りであった。

図6 前処理により作製した多孔質性の酸化物ペレット
(模擬FP含有UO2、気孔率は32%)

図7 電解還元前後の陰極内部と
還元されたペレット断面
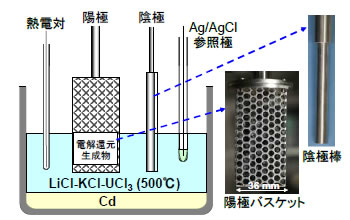
図8 電解精製槽の概略

図9 電解精製後の電極
2.3 電解還元処理におけるアクチニドとFP元素の挙動解明
2.3.1 プルトニウム(Pu)とアメリシウム(Am:代表的なMA)の溶解挙動
電解還元によりPuやAmの酸化物は金属に還元されるが、物質収支を検討するための基礎データとして、Li2Oを含む溶融LiCl中への酸化物(Pu2O3,PuO2,Am2O3,AmO2)の溶解度を測定した。溶解度はLi2O濃度の増加につれて増大したが、溶解量自体は少なく、燃料を構成する主要酸化物のPuO2では、例えば100 ℓ (約150 kg) のLiCl-1wt%Li2O中にPuは高々15 g溶解するに過ぎないことが示された。
2.3.2 アルカリ金属およびアルカリ土類金属FPの影響
使用済燃料を処理すると、溶融LiCl中にCsやSr, Baが溶解して蓄積するため、その影響を調べた。溶融LiCl中にアルカリ金属塩化物のNaClやKCl、CsClを添加した系では、Li2Oの溶解度が減少し、UO2の還元速度が大いに低下した。これは、還元反応がUO2試料内に浸透した溶融LiClを介する酸化物イオン(O2-)の拡散に律速されていることを示唆している。つまり、Li2O溶解度が大きい場合、UO2試料内外でO2-の濃度差が大きくなり、拡散によるO2-の排出が促進されるためと考えられる。一方、アルカリ土類金属塩化物のSrCl2やBaCl2では、影響は小さいことがわかった。
LiClに溶解したCsを除去することは難しいため、電解還元処理に先立って、使用済燃料からCsを出来るだけ取り除いておくことが望ましい(Csは揮発し易いため、高温処理による除去が可能)。
2.3.3 希土類元素の還元挙動
溶融LiCl中での希土類元素の挙動を電気化学測定等によって調べ、その代表としてLaの650℃でのE-pO2-図を作成した。図からは、塩中のLi2O濃度を低く維持すればLa2O3を金属にまで還元できることが示唆された。しかし熱力学的予想に反して、単独の希土類酸化物(La2O3とNd2O3)を溶融LiCl中で金属に電解還元することは実際には困難であった。一方、使用済燃料中には希土類元素と合金を形成しやすいPdなどの白金族元素FPが共存するため、2.2で述べたように、一部の希土類元素が安定な合金を生成することによって還元されることもわかった。
2.4 電解還元処理における陽極材料の検討
電解還元の陽極では、溶融塩中のO2-が酸化されてO2ガスとして排出される。従来の試験では、陽極材料として主に白金と炭素が用いられてきたが、非消耗性でO2ガスが効率良く発生する実用的な陽極を開発するために、新たな材料(金属、酸化物、ケイ素および炭素系材料)を対象とした試験を実施した。その結果、フェライトは、電気化学反応等により若干腐食するが、650℃の溶融LiCl中で安定なO2発生電極として実用できる可能性が示された。SiOは取り出せる電流密度が小さく、ホウ素ドープダイヤモンドは、500℃の溶融LiCl-KCl中ではO2発生陽極として極めて高い耐久性を有するが、より高温では良好な耐久性を示さないことが分かった。その他の材料については、溶解や不動態化が起こり、陽極として不適当であった。
3.まとめ、今後の展望
本研究開発により、乾式再処理技術が酸化物燃料に対しても適用可能であり、十分に実用化しうる技術であることを工学的観点からも実証できたと考える。現状で特に優先順位の高い開発課題としては、実規模での電解還元の実証、陽極の開発(材料と構造)が挙げられる。
乾式再処理技術は、溶融塩を用いるために臨界への裕度が大きく耐放射線性が高いこと、特別なプロセスを付加しなくてもMAがPuといっしょに回収されることなどから、特にMOX燃料や高燃焼度燃料などに対して高い経済性が期待できる。また、小規模でも経済性が保てることから、将来にわたる高速炉の建設状況に応じて柔軟に施設を増強することが可能である。今後、本研究開発を発展させることにより、乾式再処理固有の特徴を生かして、乾式再処理施設と大規模に軽水炉ウラン燃料を処理する現行のPUREX再処理施設とで上手く役割を分担し、軽水炉時代から高速炉時代への過渡期の我が国の原子燃料サイクルを支えていくシナリオを実現することができる。
4.参考文献
1) Y.Sakamura, M.Kurata and T.Inoue, J. Electrochem. Soc., 153 (3), D31-D39 (2006).
2) M.Iizuka, Y.Sakamura and T.Inoue, J. Nucl. Mater., 359, 102 (2006).
3) Y.Sakamura, T.Omori and T.Inoue, Nucl. Technol., 162, 169 (2008).