

原子力システム 研究開発事業 成果報告会資料集
分子シミュレーションによるMA含有MOX物性のモデル化に関する研究
(研究代表者)小田卓司 大学院工学系研究科 助教
1.研究開発の背景とねらい
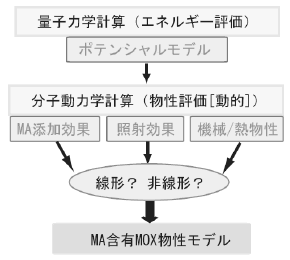
図1:計画の概要と各課題の関連性
高速増殖炉サイクルにおいて、MA含有MOX燃料の実用化は、核変換による放射性廃棄物の低減と放射能の増加による核不拡散性の向上をもたらすため、重要な研究課題として位置付けられている。しかし、MAの添加がMOX燃料の物性に与える影響については、十分な理解が得られておらず、さらなる知見の蓄積と体系化が必要である。
そこで本事業では、分子シミュレーションを用いて現象を原子スケールで分析して理解することで、MA含有MOXの機械物性、熱物性および照射応答挙動を、MAの種類と量の関数として表すモデルを構築することを目的とする。
本事業は、(1)ポテンシャルモデルの構築、(2)機械・熱物性の評価、(3)照射応答挙動の分析、の3課題から構成される。まず、量子力学計算で評価したポテンシャルエネルギー曲線を利用して、MA含有MOXに適用可能なポテンシャルモデルを構築する。そして、そのポテンシャルモデルを用いて分子動力学計算を実施することで、MA含有MOXの機械物性、熱物性ならびに照射応答挙動を、MAの種類と量の関数として評価する。その結果を体系的に整理してモデル化することで、本事業の目的が達成される(図1)。
2.研究開発成果
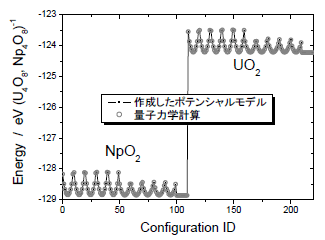
図2:作成したポテンシャルモデルと量子力学計算とのエネルギー値の比較
課題(1)の「ポテンシャルモデルの構築」においては、まず、構築する際に利用する量子力学計算の計算条件を最適化した。具体的には、UO2やPuO2、NpO2等の格子定数等を対象し、計算精度と計算時間が両立するように、k-pointサンプリング点数や内殻ポテンシャルの種類等の計算条件を検討した。UO2、PuO2、NpO2の蛍石型結晶の構造最適化計算は、密度汎関数理論に基づく量子力学計算コードであるVASPコードを利用して行った。LDA+U法を採用した場合、いずれの材料においても、格子定数について実験値と良い一致が得られた。また、GGA法においても格子定数については実験値と良い一致が得られた。一方で、LDA法やGGA+U法では、実験値との顕著な差が確認された。以上の結果から、今後の計算は主にLDA+U法、あるいはGGA法(PBE汎関数)を利用して実施することとした。内殻ポテンシャルやk-pointサンプリング点数等については、精度と速度を両立する計算条件を確認した。
最適化した計算条件の下で、VASPコードおよびCASTEPコードを利用し、UO2、PuO2、NpO2等の2酸化物、UO2-PuO2混合酸化物、およびUやPuの一部をMAで置換した系を対象として、O原子の[100]、[110]、[111]方向への変位等の基礎的な原子変位に対するポテンシャルエネルギー曲線を作成した。LDA+U法とGGA法では、ポテンシャルエネルギー曲線の傾きに明確な差が確認された。
量子力学計算を用いて多様なMA含有構造において評価したポテンシャルエネルギー曲線を利用し、原子間相互作用を記述するポテンシャルモデルを作成した。モデル式には、遠距離での収束性を良くするために指数関数を乗じた逆多項式型の2体力モデルを採用した。ポテンシャルモデル式のモデルパラメータを最適化することで、量子力学計算で評価した複数のポテンシャルエネルギー曲線を精度良く再現するモデル式を作成した(図2)。
課題(2)の「機械・熱物性の評価」においては、まず、量子力学計算を用いて体積弾性率等の評価を行った。体積弾性率については、計算値は実験値との良い一致(相違は最大で10%程度)を示した。この結果は、量子力学計算の結果をポテンシャルモデル構築に利用することが妥当であることを示唆している。ただし、課題(1)で構築したポテンシャルモデルを利用した場合、いくつかの機械物性値、特に弾性定数C44について、実験や量子力学計算の値との大きな相違が確認された。この原因は、ポテンシャルモデルとして採用した関数が不適当であること、モデル構築に利用した量子力学計算の数が不十分であること、などにあると考えられる。
課題(3)の「照射応答挙動の分析」においては、分子動力学計算による弾き出しエネルギーの評価として、UO2やMgO等の単純な金属酸化物を対象とし、弾き出しの閾値エネルギーを分子動力学計算により評価した。さらに、実験報告値と比較することで計算結果の妥当性を調べるとともに、金属酸化物において結晶構造等が弾き出し挙動に与える影響について分析した。
既往のポテンシャルモデルを利用した分子動力学計算により、UO2について弾き出しエネルギーの方位依存性を評価した結果を図3に示す。報告されている実験値と比較したところ、計算は弾き出しエネルギーを過大評価する傾向を示した。計算(0 K)と実験(室温)における温度の差が、結果に影響を与えていると考察した。また、弾き出しエネルギーの方位依存性については、UO2において同じ結晶構造を持つLi2OやCaF2と類似の挙動が得られ、結晶構造が弾き出し挙動を決定する主要な因子の一つであることが確認された。
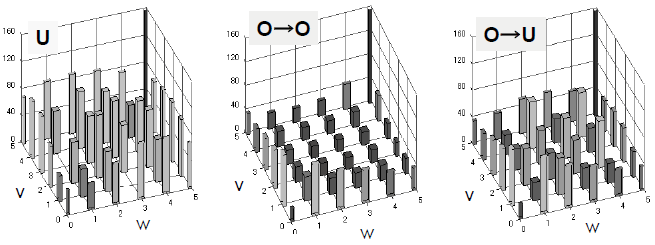
弾き出しエネルギーの温度依存性を評価するために、実験データが豊富なMgOを対象として、初期温度300 Kで弾き出しエネルギーを評価した。結果として、0 Kの場合と比べて低い弾き出しエネルギーでの欠陥生成が確認された。しかし、その温度依存性を考慮した場合においても、計算値は実験値よりも顕著に大きく、温度依存性だけでは計算における弾き出し閾値エネルギーの過大評価を説明できないことが示唆された。
3.今後の展望
構築したポテンシャルモデルの精度は、最終的に提案する材料物性評価モデルの精度に直結する。そのため、電荷変動モデル等についても検討し、より実験結果を適切に再現可能なポテンシャルモデルの構築を進める。そして、構築したモデルを利用して、弾き出しシミュレーションと機械/熱物性の評価を行う予定である。